
Driving pressure and mechanical power: new targets for VILI prevention
Introduction
Mechanical ventilation has been recognized as a cause of lung damage ever since its introduction, although the term ventilator-induced lung injury (VILI) was introduced in 1993 (1).
In Figure 1 we show the evolution of VILI since its first description. As shown, the terminology tends to reflect the cause of the injury rather than its effects. Indeed, the effects may range from micro-fractures to overt ruptures, from cytokines production to white cell clusters, from altered permeability to intra-alveolar hemorrhage. Therefore, under the term VILI we may include different pathophysiological mechanisms, each one of them with its own different pathways, ultimately leading to possibly different manifestations. In this paper, we would like to discuss the causes of VILI, how they affect the lung structures and their resulting effects. A particular focus will be devoted to the respective roles of the driving pressure and of the mechanical power to the genesis and perpetuation of VILI.

Recognized causes of VILI
Pressure
Excessive pressure leading to macroscopic rupture of lung parenchyma is the first recognized cause of ventilator lung injury and was referred to as barotrauma (2). This includes pneumothorax, pneumomediastinum, subcutaneous emphysema (6-9) and gas embolism (10). In the early 1970s, when volume controlled ventilation was mostly used and the ventilatory target was to keep the PaCO2 within a normal range, pneumothoraces were so frequent that a prophylactic insertion of bilateral chest drains was proposed to prevent death by sudden tension pneumothorax (11). The greatest fear of mechanical ventilation was not the pressure, but the high FiO2, so much so that extracorporeal membrane oxygenation (ECMO) in the first randomized controlled trial was applied to minimize FiO2 (12). With time, however, the importance of barotrauma became prominent. The level of pressure considered to be harmful had not been defined until the ARMA trial data were available (13), which suggested that a value of 30 cmH2O of airway pressure was the maximum tolerable during mechanical ventilation (14). We believe, however, that the use of a single number to define possible dangers is too simplistic, as it may lead to the administration of insufficient pressure to some patients and to excessive pressure to others. As what causes VILI is not the pressure applied to the airways, but the one applied to the lung (i.e., the transpulmonary pressure), it follows that a pressure threshold should be a value of transpulmonary pressure and not of airway pressure. Actually, the relationship between the airway pressure and the transpulmonary pressure in an individual patient is strictly linear (15). What matters, however, is the slope of such relationship, which equals the ratio of lung elastance to total respiratory system elastance (EL/Etot), which averages 0.7 in the population, but ranges from 0.2 to 0.8 (16). If we consider as a possible threshold for VILI a value of transpulmonary pressure at which some of the pulmonary units are fully inflated (i.e., in which the collagen fibers of the extracellular matrix are completely distended), a reference value of 21 cmH2O may be experimentally identified (17). In an average patient (EL/Etot =0.7), this transpulmonary pressure value would equate to 30 cmH2O airway pressure. However, in a patient in whom EL/Etot =0.8, an airway pressure of 30 cmH2O would result in an end-inspiratory transpulmonary pressure of 24 cmH2O, which corresponds to lung volume near to total lung capacity. In contrast, in a patient in whom the EL/Etot ratio is as low as 0.2 (e.g., obesity or pregnancy), the same plateau airway pressure of 30 cmH2O will correspond to a transpulmonary pressure of just 6 cmH2O, which may be associated with lung collapse and hypoxemia. Actually, a fixed airway pressure threshold of 30 cmH2O could have led in some occasions to inappropriate extracorporeal support (18), potentially avoidable if the transpulmonary pressure had been measured (19). Therefore, the plateau pressure has a meaning when it is considered in the context of the resulting transpulmonary pressure.
Volume
The focus on the importance of volume in the genesis of VILI was primarily because of Dreyfuss and colleagues who—in a series of experiments where the chest wall was wrapped to increase its elastance—showed that for VILI to occur what matters was the administered tidal volume, independently of pressures (i.e., low volume did not cause any damage even in the presence of high pressures in chest-wrapped rats, while the high volume did) (3). The long discussion between the supporters of volutrauma against the supporters of barotrauma loses any meaning if we refer to the transpulmonary pressure, instead of airway pressure, and to the strain (i.e. tidal volume normalized to the lung resting volume), instead of tidal volume. Indeed, the following relationship holds true:

i.e.,
Stress = Specific elasrance ∙ Strain [2]
Where PL is the transpulmonary pressure, VT is the tidal volume, FRC is the lung volume at atmospheric pressure, k is the lung specific elastance.
From the above relationship, it is evident that volutrauma, caused by excessive strain, is strictly connected to barotrauma, caused by excessive stress, being the specific elastance the proportionality constant (~12 cmH2O). This relationship accounts for all the results obtained by Dreyfuss in wrapped rats, a maneuver that deeply affects the airway-transpulmonary pressure relationship. The clinical relevance of tidal volume, however, has been definitely proved by the results of the ARMA trial (13), where higher tidal volume was associated with almost 10% higher mortality than lower tidal volume (12 vs. 6 mL/kg PBW).
Tidal atelectasis
The concept of atelectrauma was introduced by Arthur Slutsky and his group, after observing the sharp increase in inflammatory cytokines in in vivo experiments on rats, when the lungs were allowed to cyclically collapse and re-inflate (4). The theoretical basis for the damage induced by intra-tidal opening and closing may be found in Mead et al. (20), who discussed the stress and strain maldistribution throughout an inhomogeneous lung parenchyma. The cyclic opening and closing represents the asymptote of this phenomenon, which may also be found at a lower extent between two contiguous lung structures presenting different elasticity. A simplified approach to the Mead model is presented in Figure 2. In theory, assuming that the volume ratio between a fully expanded unit and a fully collapsed one is 10 to 1, the corresponding surface ratio, to which the stress refers, will be (10/1)2/3 =4.64. This number represents the multiplication factor of the transpulmonary pressure at the interface between the two units. Accordingly, an applied transpulmonary pressure of 30 cmH2O (leading to total lung capacity) locally results in a transpulmonary pressure (stress) equal to 30×4.64=139.2 cmH2O. It must be noted, however, that this value is purely theoretical and not experimental. Indeed, when we estimated by CT scan the stress and strain maldistribution, we found that the multiplication factor in a pathologically inhomogeneous lung is about 2 in about 40% of the ventilatable lung parenchyma (21). This means that a transpulmonary pressure of 15 cmH2O may be locally as high as 30 cmH2O, i.e., more than enough to reach the maximal lung capacity, a physical limit to lung expansion with possible devastating effect (17).

Flow
The possible effect of excessive gas flow on pulmonary injury has attracted less attention than the factors listed so far. However, both theoretical considerations (22,23) and experimental evidence (24) suggest that during mechanical ventilation the importance of flow cannot be neglected. Indeed, the flow may be considered as the rate at which a given strain occurs into the lung. As the lung parenchyma roughly behave as a viscoelastic body, the higher the rate of strain, the greater the resistance developing within the extracellular matrix. This process requires energy, which is proportional to the rate of strain and is dissipated into the lung parenchyma. Although this is an oversimplification of the phenomena occurring throughout the lung parenchyma, it accounts reasonably well for the experimental observations. When the energy dissipated into the lung parenchyma was measured as the change in pressure after the inspiratory flow has been suddenly interrupted (a phenomenon called ‘stress relaxation’) (25), it was found that the dissipated energy increases with respiratory rate and lung inhomogeneity (26). Therefore, a given lung strain may or may not result in lung injury depending on the rate at which it develops. Unfortunately, we do not know exactly if there is harmful threshold of flow, but this should be considered anyway in the framework of the other possible causes of VILI.
Respiratory rate
The effects of the respiratory rate on VILI are so intuitively obvious that it is surprising to realize how little attention has been paid to it. Unquestionably, if a given tidal volume is dangerous at a rate of 15 bpm, one might expect that it would be more dangerous at 30 bpm. Indeed, the effects of respiratory rate on VILI have been described in experimental animals (27,28). In particular, when we applied a strain greater than 2, which invariably leads to death when delivered at a rate of 15 bpm, we found a lack of injurious effect if delivered at 3 or 6 bpm (29). The relevance of respiratory rate underlines another possible scenario: indeed, in analogy with materials fatigue, it may be possible that damage occurs only after a given number of stress and strain cycles have been delivered and generated into the lung (i.e., VILI below the stress at rupture requires time).
Interaction between ventilator and lung parenchyma
Mechanical interaction
All the above-mentioned factors may lead to VILI, but it is worth discussing which are the physical and biological bases for VILI.
Physical
The physical basis of VILI is represented by a broad spectrum of possible insults, starting from an excessive deformation of the extracellular matrix, to micro-fractures in its structure, up to frank stress-at-rupture. Either one of these physical insults can trigger, to different extent, an inflammatory response. The quantification of these processes, however, is surprisingly lacking. Intuitively, below a given threshold, stress and strain are well tolerated while, beyond that, the sequence ‘unphysiological deformation—micro-fracture—stress-at-rupture’ develops. Each of these processes requires increasing amounts of energy. The hyaluronan, differently from collagen and elastin, may represent one of the weaker load-bearing structures of the extracellular matrix in the lung. Although, we do not know the ‘quantum’ of energy sufficient to break the molecular bonds of such molecule, its order of magnitude shouldn’t be very different from the force necessary to break the molecular bond between hyaluronic acid and its binding protein, which averages ~(40±11)×10-12 Newtons (30). Given a displacement of 2.8×10-6 m, the ‘quantum’ of energy would be ~1.12×10-16 J. At an average molecular weight of hyaluronans of 2,500 kDa, if we accept for humans an amount of hyaluronan of ~0.1×10-6 g/g of lung tissue (31), we may speculate on the relationship between the energy input (from the ventilator) and the likelihood of molecular breakage. An energy per breath of ~0.5 J would correspond to an average amount of energy/molecule between 10% and 30% of the energy required for breaking (the above mentioned energetic ‘quantum’). Assuming that the energy required to rupture the hyaluronan molecules follow a normal statistical distribution, it is very likely that few molecules may break at every cycle and they will undergo repair (31). If the energy/molecule increases, either because the greater amount of delivered energy or because of the maldistribution of forces due to increased inhomogeneity, the rate of fracture will increase. If the rate of fracture exceeds the capability of physiological repair, with time VILI would manifest.
Biological
Although the trigger for VILI must necessarily be mechanical in its nature, the inflammatory reaction that follows excessive deformation or micro-fractures plays a major role. The first reaction is the production of cytokines, originating either from abnormally distorted epithelial cells or from hyaluronan fragments which will trigger a toll-like receptors (TLRs) mediated inflammatory reaction (32,33). Once the inflammatory reaction is fully activated, the consequences are the typical ones: increased vascular permeability, inflammatory cell migration, increased platelets adhesion, activation of the tissue-factor pathway, etc. All these processes lead to a profound remodeling of the extracellular matrix, increasing its degradation and maintaining the inflammatory stimulus. On the other hand, it must be reminded that the inflammatory reaction is also necessary to drive lung repair, whose mechanisms and possible interactions with mechanical ventilation are to date largely unknown.
Damage manifestations
Gas leak
VILI has different macroscopic and microscopic manifestations. The first recognized manifestations of VILI were due to stress-at-rupture, leading to gas entry in different districts or cavities. These manifestations depend on the interaction between the ventilation and the lung parenchyma. Although we observed the occurrence of pneumothorax during total lung rest during ECMO due to tissue necrosis, most commonly VILI occurs in the more ‘healthy’ regions of the lung which can still be ventilated. These regions are somehow protected from VILI because of surfactant deficit and early fibrosis. The presence of these two factors, for a given pressure, results in lower strain. Nevertheless, lung pathology undoubtedly determines two other conditions that increase the likelihood of VILI: reduced lung size and lung inhomogeneity.
Altered permeability and interstitial edema
If stress and strain are so elevated to induce stress-at-rupture, the air leak will immediately follow without any other microscopic features. However, if stress and strain are pathologically elevated but insufficient to cause alveolar rupture, the manifestations observed are mostly related to the inflammatory cascade. For this to occur, however, time is required. In animal ventilated at 15 bpm with a tidal volume greater than twice the FRC (i.e., lung strain greater than 2), we observed the first lesions only after several hours (Figure 3). These lesions were small spots of increased CT density along the visceral pleura (stress raiser). After their appearance, the process accelerated exponentially to complete end-expiratory lung collapse/edema. Interestingly, these VILI-related lung alterations were fully recruitable during inspiration (34). These observations are consistent with the following sequence of events: (I) mechanical stimuli trigger inflammation through micro-fractures and distortions; (II) capillary permeability increases with interstitial edema; (III) the pulmonary units are compressed but recruitable. Obviously other accompanying phenomena may be observed, as capillary micro-fractures and alveolar/capillary walls ruptures (35). It must be noted, however, that for edema to occur, even in the presence of increased capillary permeability, a sufficient blood flow is required. Therefore, a reduction in cardiac output due to high PEEP, may decrease edema formation. We wonder whether some of the ‘protective effects’ of PEEP could be simply due to preventing the manifestations of the damage (i.e., edema) rather than the damage itself (inflammation and altered permeability).

The ‘new entries’
Driving pressure
The driving pressure (i.e., plateau pressure minus PEEP) is currently considered the best predictor of VILI in patients with ARDS (36) as well as in patients undergoing general anesthesia, in whom the rate of pulmonary complications was associated with higher levels of driving pressure (37). Unfortunately, although very fashionable, the evidence behind driving pressure is at best indirect and derives from statistical models of retrospective studies. Indeed, it is worth noting that:
- The original evidence in support of driving pressure—as a mediator of mortality—derives from a series of studies which were retrospectively analyzed through a very sophisticated statistical procedure (i.e., mediation analysis). The multilevel ‘causal’ mediation analysis of individual trial data showed that a reduction in driving pressure post randomization is an independent variable (mediator) associated with survival. Although both randomization (i.e., the allocation to the lower VT treatment group) and a change in mediator (a reduction in driving pressure) had a significant effect on survival, when analyzed together only a reduction in driving pressure -not randomization- independently explained the survival benefit (complete mediation). In other words, a reduction in driving pressure ‘mediated’ most of the effects attributed to randomization with survival that exceeded the main effect of treatment group allocation.
However, ‘causal mediation’ does not establish a direct causal link between setting a specific driving pressure and outcome, as driving pressure is mathematically coupled with tidal volume and elastance. Therefore, a change in elastance, which may follow an intervention, indicates a change in lung mechanics which transcends the setting of a specific value of driving pressure. When plateau pressure, tidal volume and PEEP are set within the tight ranges of a protective ventilation, driving pressure per se may not offer any additional advantage over the indices of lung mechanics such as elastance, compliance or plateau pressure as recently shown by Guerin et al. (38). Indeed, independently from sophisticated statistics, the best association between outcome and driving pressure instead of tidal volume/kg IBW is self-evident considering the relationship between the two variables:
∆P = VT × E
As shown, the influence of driving pressure on outcome may be either due to elastance (severity of the disease) or tidal volume (degree of strain).
- The major drawback of using driving pressure in isolation is that it doesn’t take into account the role of PEEP. For example, a theoretically ‘safe’ level of driving pressure of 12 cmH2O could become harmful if PEEP is 20 or 0 cmH2O, depending on the clinical condition. Finally, as for other mechanical parameters, it is worth noting that the driving pressure should be referred to the lung and not the respiratory system.
Mechanical power
In a series of experiments on healthy animals we found that a strain considered lethal (i.e., greater than 2) was only fatal when delivered at 15 breaths per minute (39), but not if delivered at rates of 3 to 6 bpm (29). In addition, we found that the tidal strain was more related to injury than the static strain (40). Further, we found that, for the same tidal volume, the rate of its delivery (i.e., the flow) was also a possible determinant of VILI (24). Given these experimental data, we realized that the cause of VILI could be described as a single physical entity (i.e., the mechanical power), which combines volume, pressures, flow and respiratory rate (22). Indeed, if we consider the classical equation of motion (41), which quantifies all the different pressures present in the respiratory system at any given moment, and we multiply that total pressure by the changes in lung volume and rate, we obtain the following power equation:
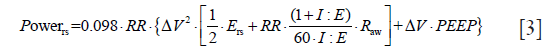
Where 0.098 is the conversion factor from L × cmH2O to J, RR is the ventilatory rate, Ers is the elastance of the respiratory system, I:E is the ratio between inspiratory and expiratory time, Raw is the airway resistance, ∆V is the tidal volume. Accordingly, in a normal subject breathing at 15 bpm, with a tidal volume of 0.5 L with an I:E of 1:1, a normal respiratory system elastance of 10 cmH2O/L and Raw of 10 cmH2O/L/s at 0 PEEP, the mechanical power in J/min equals 3.675.
To fully understand this equation, it is helpful to consider each component separately, starting from the equation of motion:
P= Ers ∙ ∆V + Raw ∙ F + PEEP [4]
Where P is the pressure in the respiratory system at any given moment, Ers is the total elastance of the respiratory system, ∆V is the tidal volume, Raw are the airway resistances, F is the flow and PEEP is the positive end-expiratory pressure.
As shown, all the components of VILI are represented:
- The product Ers × ∆V, which is the pressure needed to overcome the elastic forces of the whole respiratory system, is nothing else than the driving pressure we discussed above;
- The product Raw × F is the pressure needed to move the gas inside the respiratory system;
- PEEP is the pressure generating the baseline static stretch of the lung fibers.
The relevance of each component is immediately evident examining the graphical representation of the equation of power (Eq. 3 and Figure 4). The introduction of flow in this equation as well as the introduction of PEEP deserves a discussion. Indeed, the pressure due to the flow is usually considered fully dispersed within the airways, while the pressure at end-expiration is usually considered protective and not taken into account for the calculation energy during tidal breathing.

Flow
Although the energy linked to the flow is primarily dissipated throughout the airways, the flow rate affects the stress relaxation (i.e., the change of pressure when the constant volume is maintained) (25). The stress relaxation is due to a possible pendelluft phenomenon and to the energy dissipated throughout the parenchyma because of shear forces into the extracellular matrix. Therefore, we believe that the flow component cannot be ignored.
PEEP
PEEP is traditionally considered protective against VILI and this belief originated from the first seminal observations by Webb and Tierney (42). However, the protective effect of PEEP primarily manifests when it is associated with a decrease in tidal volume. The important role of PEEP in the mechanical power should become clearer if we consider the physical characteristics of an elastic system. The force needed to extend an elastic structure is directly proportional to the extent of displacement, as shown by Hook’s law:
F = k ∙ x [5]
Where F is the force, x is the displacement and k a constant related to the intrinsic characteristics of the system. From that, it is clear how the force (and so the energy) needed to displace an elastic body (i.e., the lung) strictly depends on the degree of displacement from resting condition (FRC) already present in the system at baseline (at end-expiration). Therefore, the applied level of PEEP multiplied by the tidal volume (see Figure 4) represents the energy level to be overcome to generate each tidal volume. In fact, the energy is not the product of the variation of pressure multiplied by the change in volume, but the product of the absolute pressure multiplied by the change in volume. This is the reason why it is correct to include PEEP in the equation of power.
A key challenge for the application of mechanical power is that it needs to be adapted to account for the differences in lung size, degree of inhomogeneity and local distribution of stress and strain. In other words, the same mechanical power may produce different effects in healthy or injured lungs.
Conclusions
The difference between the use of driving pressure or mechanical power as potential predictors or markers of VILI are mathematically, physiologically and conceptually evident as driving pressure is just one of the cases of VILI which are included in the equation of mechanical power. However, the clinical application of both mechanical power and driving pressure lack the direct proof. What is needed is a way to normalize the mechanical power to the lung size and the degree of inhomogeneity. A threshold of mechanical power could guide on the proper use of mechanical ventilation or on the application of extracorporeal respiratory assistance.
Acknowledgements
None.
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Parker JC, Hernandez LA, Peevy KJ. Mechanisms of ventilator-induced lung injury. Crit Care Med 1993;21:131-43. [Crossref] [PubMed]
- Kumar A, Pontoppidan H, Falke KJ, et al. Pulmonary barotrauma during mechanical ventilation. Crit Care Med 1973;1:181-6. [Crossref] [PubMed]
- Dreyfuss D, Soler P, Basset G, et al. High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis 1988;137:1159-64. [Crossref] [PubMed]
- Tremblay L, Valenza F, Ribeiro SP, et al. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest 1997;99:944-52. [Crossref] [PubMed]
- Marini JJ, Jaber S. Dynamic predictors of VILI risk: beyond the driving pressure. Intensive Care Med 2016;42:1597-600. [Crossref] [PubMed]
- Zimmerman JE, Dunbar BS, Klingenmaier CH. Management of subcutaneous emphysema, pneumomediastinum, and pneumothorax during respirator therapy. Crit Care Med 1975;3:69-73. [Crossref] [PubMed]
- de Latorre FJ, Tomasa A, Klamburg J, et al. Incidence of pneumothorax and pneumomediastinum in patients with aspiration pneumonia requiring ventilatory support. Chest 1977;72:141-4. [Crossref] [PubMed]
- Woodring JH. Pulmonary interstitial emphysema in the adult respiratory distress syndrome. Crit Care Med 1985;13:786-91. [Crossref] [PubMed]
- Gammon RB, Shin MS, Buchalter SE. Pulmonary barotrauma in mechanical ventilation. Patterns and risk factors. Chest 1992;102:568-72. [Crossref] [PubMed]
- Marini JJ, Culver BH. Systemic gas embolism complicating mechanical ventilation in the adult respiratory distress syndrome. Ann Intern Med 1989;110:699-703. [Crossref] [PubMed]
- Hayes DF, Lucas CE. Bilateral tube thoracostomy to preclude fatal tension pneumothorax in patients with acute respiratory insufficiency. Am Surg 1976;42:330-1. [PubMed]
- Zapol WM, Snider MT, Hill JD, et al. Extracorporeal membrane oxygenation in severe acute respiratory failure. A randomized prospective study. JAMA 1979;242:2193-6. [Crossref] [PubMed]
- ARDS Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med 2000;342:1301-8. [Crossref] [PubMed]
- Tobin MJ. Culmination of an era in research on the acute respiratory distress syndrome. N Engl J Med 2000;342:1360-1. [Crossref] [PubMed]
- Cressoni M, Chiumello D, Algieri I, et al. Opening pressures and atelectrauma in acute respiratory distress syndrome. Intensive Care Med 2017;43:603-11. [Crossref] [PubMed]
- Chiumello D, Carlesso E, Cadringher P, et al. Lung stress and strain during mechanical ventilation for acute respiratory distress syndrome. Am J Respir Crit Care Med 2008;178:346-55. [Crossref] [PubMed]
- Protti A, Andreis DT, Milesi M, et al. Lung anatomy, energy load, and ventilator-induced lung injury. Intensive Care Med Exp 2015;3:34. [Crossref] [PubMed]
- Australia New Zealand Extracorporeal Membrane Oxygenation Influenza Investigators, Davies A, Jones D, et al. Extracorporeal Membrane Oxygenation for 2009 Influenza A(H1N1) Acute Respiratory Distress Syndrome. JAMA 2009;302:1888-95. [Crossref] [PubMed]
- Grasso S, Terragni P, Birocco A, et al. ECMO criteria for influenza A (H1N1)-associated ARDS: role of transpulmonary pressure. Intensive Care Med 2012;38:395-403. [Crossref] [PubMed]
- Mead J, Takishima T, Leith D. Stress distribution in lungs: a model of pulmonary elasticity. J Appl Physiol 1970;28:596-608. [PubMed]
- Cressoni M, Cadringher P, Chiurazzi C, et al. Lung inhomogeneity in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 2014;189:149-58. [PubMed]
- Gattinoni L, Tonetti T, Cressoni M, et al. Ventilator-related causes of lung injury: the mechanical power. Intensive Care Med 2016;42:1567-75. [Crossref] [PubMed]
- Marini JJ. Strain Rate and Cycling Frequency-The "Dynamic Duo" of Injurious Tidal Stress. Crit Care Med 2016;44:1800-1. [Crossref] [PubMed]
- Protti A, Maraffi T, Milesi M, et al. Role of Strain Rate in the Pathogenesis of Ventilator-Induced Lung Edema. Crit Care Med 2016;44:e838-45. [Crossref] [PubMed]
- Milic-Emili J. Pulmonary flow resistance. Lung 1989;167:141-8. [Crossref] [PubMed]
- Ganzert S, Moller K, Steinmann D, et al. Pressure-dependent stress relaxation in acute respiratory distress syndrome and healthy lungs: an investigation based on a viscoelastic model. Crit Care 2009;13:R199. [Crossref] [PubMed]
- Vaporidi K, Voloudakis G, Priniannakis G, et al. Effects of respiratory rate on ventilator-induced lung injury at a constant PaCO2 in a mouse model of normal lung. Crit Care Med 2008;36:1277-83. [Crossref] [PubMed]
- Retamal J, Borges JB, Bruhn A, et al. High respiratory rate is associated with early reduction of lung edema clearance in an experimental model of ARDS. Acta Anaesthesiol Scand 2016;60:79-92. [Crossref] [PubMed]
- Cressoni M, Gotti M, Chiurazzi C, et al. Mechanical Power and Development of Ventilator-induced Lung Injury. Anesthesiology 2016;124:1100-8. [Crossref] [PubMed]
- Liu X, Sun JQ, Heggeness MH, et al. Direct quantification of the rupture force of single hyaluronan/hyaluronan binding protein bonds. FEBS Lett 2004;563:23-7. [Crossref] [PubMed]
- Fraser JR, Laurent TC, Laurent UB. Hyaluronan: its nature, distribution, functions and turnover. J Intern Med 1997;242:27-33. [Crossref] [PubMed]
- Jiang D, Liang J, Fan J, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 2005;11:1173-9. [Crossref] [PubMed]
- O'Neill LA. TLRs play good cop, bad cop in the lung. Nat Med 2005;11:1161-2. [Crossref] [PubMed]
- Cressoni M, Chiurazzi C, Gotti M, et al. Lung inhomogeneities and time course of ventilator-induced mechanical injuries. Anesthesiology 2015;123:618-27. [Crossref] [PubMed]
- West JB. Invited review: pulmonary capillary stress failure. J Appl Physiol (1985) 2000;89:2483-9; discussion 97. [PubMed]
- Amato MB, Meade MO, Slutsky AS, et al. Driving pressure and survival in the acute respiratory distress syndrome. N Engl J Med 2015;372:747-55. [Crossref] [PubMed]
- Serpa Neto A, Schmidt M, Azevedo LC, et al. Associations between ventilator settings during extracorporeal membrane oxygenation for refractory hypoxemia and outcome in patients with acute respiratory distress syndrome: a pooled individual patient data analysis: Mechanical ventilation during ECMO. Intensive Care Med 2016;42:1672-84. [Crossref] [PubMed]
- Guérin C, Papazian L, Reignier J, et al. Effect of driving pressure on mortality in ARDS patients during lung protective mechanical ventilation in two randomized controlled trials. Crit Care 2016;20:384. [Crossref] [PubMed]
- Protti A, Cressoni M, Santini A, et al. Lung stress and strain during mechanical ventilation: any safe threshold? Am J Respir Crit Care Med 2011;183:1354-62. [Crossref] [PubMed]
- Protti A, Andreis DT, Monti M, et al. Lung stress and strain during mechanical ventilation: any difference between statics and dynamics? Crit Care Med 2013;41:1046-55. [Crossref] [PubMed]
- Rodarte JR, Rehder K. Dynamics of respiration. In: Macklem PT, Mead J, editors. Handbook of Physiology. Baltimore: Williams & Wilkins; 1986:131-44.
- Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis 1974;110:556-65. [PubMed]

