

Lessons learned in acute respiratory distress syndrome from the animal laboratory
Challenges in modeling acute respiratory distress syndrome (ARDS)
ARDS in humans constitutes acute hypoxemic respiratory failure after an inciting insult caused by a neutrophil-rich exudative pulmonary edema (1-3). Associated features include microvessel thrombi, increased permeability across both endothelial and alveolar epithelial layers, reduced fluid clearance, and protein-rich hyaline membrane formation (4,5). Subsequently, the diffuse alveolar damage (DAD) of the acute phase will either gradually resolve, or progress as interstitial fibrosis (6), leading to pulmonary impairment in survivors (7).
The fundamental mechanisms responsible for ARDS remain elusive, despite over 50 years of research. The syndrome remains defined clinically, without reliable biochemical or molecular diagnostics. Patient heterogeneity remains the major limitation to using humans to investigate the mechanisms underlying ARDS, although significant advancements have been made in understanding ARDS epidemiology (3,8,9) and in testing specific treatments and modalities of supportive care (10-19). As of now, there are no proven successful specific treatments for ARDS, and supportive care using protective ventilation limiting tidal volumes (VT) (10) and plateau pressures (PPlat) (9), using some amount of positive end-expiratory pressure (PEEP) (20), restrictive fluid balances (11), and neuromuscular blockade (NMB) (15), prone positioning (16), or extracorporeal membrane oxygenation for refractory cases (17).
Animal models provide an accepted pre-clinical platform for testing therapies and understanding pathophysiology in human disorders. Animal experiments bridge clinical research and basic bench science. Animal models progress from simplicity, efficiency, and uncertain applicability to humans (rodents), to significant complexity, expense, and translatability (primates). No single animal model captures all of the features of human ARDS, making pre-clinical modeling and testing of therapies difficult. This likely also affects the poor translatability of interventions tested in animal models to human ARDS. The poor translatability is exacerbated by a lack of clarity in whether the major causes of injury are due to the underlying insult, or the associated mechanical ventilation. The limitations of animal models of ARDS have been extensively reviewed (21).
Nevertheless, specific models of acute lung injury (ALI) (here used exclusively to refer to animal models) do capture elements of human ARDS (Table 1). Interpreted within the limits of the model and the specific interventions being tested, animal models can provide excellent targeted data regarding specific aspects of ARDS physiology. These findings also need to be interpreted in light of the specific model organism. For example, rodent models have an exaggerated response to produce nitric oxide (22) and recognize different pathogen-associated molecular patterns at different affinities due to differences in their Toll-like receptors (23). Sheep possess intravascular pulmonary macrophages which predispose them to worse lung injury after endotoxin challenge (24). However, mice have the advantage of (relatively) easily created knockouts and other genetic manipulations to interrogate pathways, whereas larger animals allow for greater physiologic monitoring and longer lengths of ventilation more congruent with human ARDS. An official American Thoracic Society statement on experimental ALI (25) suggested that animal models should reflect at least three of four central features: histological evidence of injury, barrier disruption (evidence of edema), inflammation (neutrophil and cytokine accumulation), and physiologic dysfunction (usually gas exchange). While acknowledging that not every model system will be able to report all requested parameters, these guidelines were placed to ensure some consistency and assurance that the injury being modeled is, in fact, comparable to human ARDS.

Full table
Ventilator-induced lung injury (VILI)
The single most translatable finding from animal models of ARDS was the damage associated with mechanical ventilation, a multifactorial entity referred to as VILI. Mechanical ventilation at excessive VT or pressures leads to pathophysiological changes reminiscent of human ARDS (26,27). Damage was shown to be both magnitude- and duration-dependent (27-30), with both excessive stretch (27,28) and atelectasis implicated (31). Additionally, VILI was shown to be influenced by animal size, whether the thorax was open or closed, and whether extrinsic PEEP was used.
In 1974, Webb and Tierney subjected intact rats to mechanical ventilation up to peak pressures up to 45 cmH2O, and demonstrated that higher pressures led to worse and faster pulmonary edema (32), unequivocally demonstrating the potential for VILI. Further studies examining vascular pressures and filtration subsequently clarified that this edema was not primarily hydrostatic, but rather, represented loss of barrier function, more consistent with inflammatory exudate (33-36).
Different animal models have demonstrated different thresholds for what constitutes injurious ventilation. VT >30 mL/kg are required for stretch-related injury in rodent models, with any VILI visible in rodents ventilated with smaller VT likely due to atelectasis, readily resolved with a recruitment maneuver (37). This issue is confounded by the difficulties in ventilating smaller animals for extended time periods as they are prone to temperature and hemodynamic instability, with less invasive monitoring. Therefore, given the shorter time-courses of ventilation in small animal experiments, larger VT is employed. In a meta-analysis of animal studies of VILI including mice, rats, rabbits, pugs, and sheep, Caironi et al. proposed that maximal lung strain, defined as 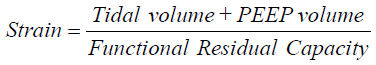 was the parameter most associated with time to VILI, and served as a unifying principle for differences in VILI generation among different animal species (38). These authors also identified an apparent threshold for strain >2 as associated with near-immediate VILI. Maximal lung stress (transpulmonary pressure at end-inspiration) was also associated with time to VILI generation, although not as strongly as was strain.
was the parameter most associated with time to VILI, and served as a unifying principle for differences in VILI generation among different animal species (38). These authors also identified an apparent threshold for strain >2 as associated with near-immediate VILI. Maximal lung stress (transpulmonary pressure at end-inspiration) was also associated with time to VILI generation, although not as strongly as was strain.
Animal evidence about the risks of VILI was translated into clinical practice after the publication of the landmark ARDSNetwork trial comparing low VT [6 mL/kg ideal body weight (IBW), PPlat ≤30 cmH2O] versus high VT (12 mL/kg IBW, PPlat ≤50 cmH2O), showing a 9% absolute mortality reduction with lower VT. This study was limited to effectively using VT and airway PPlat as surrogates for strain and stress, respectively. However, these may not be the best estimates of strain and stress, and one size may not fit all patients, particularly in a heterogeneous process like ARDS. Ongoing studies, discussed in more detail below, have taken advantage of imaging to further quantify the heterogeneity of VILI in animal models of ALI, and attempt to move us beyond VT and airway pressures.
In a separate line of VILI investigations, extrinsic PEEP has also been tested in animal models of ALI, with conflicting results. When peak pressures and end-inspiratory volumes are kept constant, PEEP has generally been protective in animal models (32,39,40), with suggestion that large cyclic changes in lung volume promote edema formation, with protection by PEEP. The reduction in cardiac output in response to PEEP in closed-chest animals was also thought to contribute to lower edema formation with PEEP (41,42). Thus, in healthy lungs subject to injurious ventilation, the beneficial effects of PEEP appear attributable to reducing cyclic deformation, reduced cardiac output, and potentially surfactant stabilization. However, the generalization of this to humans, particularly critically ill humans, is questionable. Reduction in cardiac output, for example, can be deleterious for patients with shock and already compromised oxygen delivery.
In an acid aspiration model of ALI in rats, PEEP prevented worsening inflammation and edema (43). In a model of surfactant depletion, significant atelectasis contributed to worsened oxygenation and over-distension in non-atelectatic lung, which was reversed by application of PEEP and surfactant (44). In a separate study using direct in vivo microscopy, investigator exposed surfactant-depleted porcine ALI model to different combinations of VT and PEEP, and demonstrated that a low VT (6 mL/kg) and high PEEP (20 cmH2O) resulted in the optimal stabilization of alveoli, relative to higher VT and lower PEEP levels (45). These results need to be considered in light of the limitations of the surfactant depletion model (Table 1): specifically, the responsiveness of this particular model (of de-recruitment) to PEEP (re-recruitment).
However, the relevance of these findings to human ARDS is less clear. To date, four trials of high versus low PEEP have failed to demonstrate an advantage to higher PEEP (18,19,46,47). While oxygenation improved, this did not translate to improved mortality, and in at least one instance, aggressive recruitment and setting PEEP according to best compliance resulted in increased mortality (47). This is potentially related to animal models of ALI failing to reproduce the frailty and co-morbidities of human ARDS. Experiments of PEEP in healthy animals may underestimate the cardiovascular effects of PEEP and recruitment maneuvers in critically ill adult humans with ARDS. This is particularly salient when we recognize that most deaths in ARDS are due to multisystem organ failure or withdrawal of care due to poor neurologic or medical prognosis, rather than hypoxemia (48,49). Thus, the animal models of PEEP in ALI may have appropriately translated the benefits of PEEP on re-recruitment, alveolar stabilization, and hypoxemia, but failed to translate the hemodynamic risks.
Imaging to gain insight into ARDS and mechanical ventilation
While global reductions in VT and airway PPlat have occurred after publications demonstrating improved outcomes with lower VT ventilation, concerns about VILI persisted, but became significantly more nuanced. Regional heterogeneity was appreciated as a potential contributor to ongoing VILI despite low VT. Advanced imaging techniques in animal models of ALI have allowed us to quantify these observations and develop testable hypotheses regarding residual contributors to VILI.
Regional airspace sizes can be estimated in vivo during mechanical ventilation by measuring the apparent diffusion coefficient (ADC) of helium-3 (3He) using diffusion-weighted hyperpolarized gas magnetic resonance imaging (MRI) (44,50,51). A study of surfactant-depleted rats demonstrated the co-existence of atelectasis and over-distension, consistent with atelectasis resulting in reciprocal overdistension of neighboring airspaces, with improvement after the addition of PEEP and exogenous surfactant (44). These pockets of atelectasis and overdistension were not visible on computed tomography (CT), confirming the utility of modalities such as hyperpolarized gas MRI. Significantly, overdistension was not confined to non-dependent lung regions, but occurred within the atelectatic regions, suggesting that the reciprocal overdistension was occurring within neighboring airspaces, and was a local as well as a global phenomenon. Finally, overdistension occurred in this model despite using VT of 10 mL/kg, lower than what is typically used to exacerbate VILI in surfactant-depleted rats. These findings were confirmed using synchrotron CT to obtain Xenon-enhanced ventilation and density maps in surfactant-depleted rabbits (52). These studies have contributed to our understanding of VILI by demonstrating that atelectasis and overdistension can co-exist in close proximity.
In a separate series of experiments, investigators used CT to assess how lung injury was propagated in the presence or absence of prior injury (53). In the absence of an inciting injury, rats were ventilated with injurious (30 mL/kg) VT and zero PEEP. Injury started in the periphery, and moved centrally towards the hilum in this VILI model. Importantly, all animals experienced strain >2. As a comparator, a separate group of rats were exposed to acid aspiration and modest (12 mL/kg) VT and low (3 cmH2O) PEEP. In this model with pre-existing lung injury, two patterns of injury from acid aspiration were noted: propagated and contained. When examining which variables predicted propagation of injury, the strongest predictor was strain, with a strain >1.29 associated with injury propagation and mortality. Interestingly, hypoxemia after initial acid aspiration did not predict whether injury would propagate or stay contained. In the animals with propagating injury, injury progressed from an initial central distribution to the periphery, the opposite pattern than that of VILI in uninjured rats. This study, for the first time, documented the progression of different types of lung injury, reinforced the importance of strain for predicting injury propagation, and quantified the lower threshold of excess strain necessary for injury propagation in the setting of a pre-existing inflammatory insult.
Positron emission tomography (PET) has also been used to understand pulmonary physiology, as well as inflammation during experimental ALI. Imaging of surfactant-depleted sheep after injection of nitrogen-13 (13N2) demonstrated a consistent inverse correlation between regional shunt and regional aeration (54). Interestingly, in response to PEEP, while aeration (expectedly) improved, the change in perfusion was more variable, visually demonstrating the difference between PEEP-induced gains in aeration (anatomic recruitment) and the ensuing contribution of that region to the total shunt fraction and oxygenation (functional recruitment). These relationships were only evident using imaging and assessing regional contributions, as the association between global aeration and global shunt fraction was poor. Thus, the physiologic responses to PEEP (oxygenation) appear to be dissociated from anatomic recruitment, related to differential effects on aeration and blood flow. In a separate study (55), this group demonstrated using 13N2-PET that prone positioning improves gas exchange in surfactant-depleted sheep by improving aeration and thus reducing shunt in the dorsal regions of the lung. Blood flow to the dorsal regions remained substantial; thus, the mechanism of improved oxygenation was likely reduced shunt fraction, rather than re-direction of blood flow away from atelectatic regions. The authors also noted that recruitment of dorsal lung spaces led to a more homogenous distribution of VT and lower airway pressures, suggesting beneficial effects related to minimizing VILI while in the prone position.
PET scans tracking uptake of 18F-fluorodeoxyglucose have demonstrated neutrophil accumulation and activation in experimental ALI (56-58). However, PET requires a substantial radiation burden and long acquisition times. Novel imaging methodologies exploit the accumulation of lactate and elevated lactate-to-pyruvate ratio (LPR) as surrogate markers for neutrophil accumulation (43,59). Hyperpolarized carbon-13 (13C) MRI tracks the fate of injected 13C-pyruvate during ALI, allowing for identification of the regional LPR, and presumably inflammation. In an acid aspiration rat model undergoing low (8 mL/kg) VT ventilation with (5 cmH2O) or without (0 cmH2O) PEEP, hyperpolarized 13C MRI demonstrated elevated LPR predominantly in the dependent lungs of animals ventilated without PEEP (43). LPR was correlated with hyaline membrane deposition, edema formation, and expression of myeloperoxidase, consistent with its role as a non-invasive biomarker of neutrophil activity. This study contributes additional imaging evidence that atelectasis contributes to lung inflammation, and that this inflammation can be mitigated by PEEP. However, it should be noted that in separate studies comparing different doses of acid aspiration, LPR was lower in rats exposed to higher acid concentrations, a phenomenon partially explained by hyper-perfusion of those regions (59). Thus, 13C MRI needs to be interpreted in light of other metrics of ALI, such as oxygenation and compliance, to be useful.
Spontaneous breathing during mechanical ventilation
One of the ongoing controversies regarding mechanical ventilation and minimizing risk of VILI deals with whether, to what degree, and in which patients should be allow spontaneous respiratory effort. Spontaneous ventilation has been described as both beneficial and harmful in animal models of ALI. In healthy rabbits subject to modest (10 to 15 mL/kg) VT and low (2 cmH2O) PEEP, spontaneous breathing lowered PaCO2 and reduced dead space (60). However, in surfactant-depleted rabbits, moderate (7 to 9 mL/kg) VT with strong spontaneous effort had worse lung injury compared with lower (6 mL/kg) VT or less spontaneous breathing (61). The least lung injury occurred in animals with low (6 mL/kg) VT and strong spontaneous breathing. In these studies, all animals had moderate (8 cmH2O) levels of PEEP. These authors concluded that the benefits of spontaneous breathing during mechanical ventilation after surfactant-depletion depended upon the VT used. In the group with moderate (7 to 9 mL/kg) VT with strong spontaneous effort (highest damage), transpulmonary pressures often exceeded 30 cmH2O, despite airway PPlat remaining <30 cmH2O, whereas this threshold was not breached in animals with low (6 mL/kg) VT and strong spontaneous breathing (least damage). Thus, as long as spontaneous effort was not causing excessive transpulmonary pressures, it may have some benefit by recruiting dependent lung.
These same authors extended these findings to testing the benefits of spontaneous breathing on two different severities of lung injury (62). Mild ALI was induced by surfactant depletion, and severe ALI was induced by surfactant lavage followed by injurious ventilation for two hours. Rabbits were then subject to controlled mechanical ventilation with and without strong spontaneous effort at modest (9 to 11 cmH2O) PEEP. In mild ALI, spontaneous breathing was beneficial, whereas in severe ALI, spontaneous breathing worsened lung injury, with higher transpulmonary pressures than the other groups, consistent with the previous studies. Thus, in severe ALI, spontaneous breathing has more potential to result in excessive transpulmonary pressures, and should be avoided. This was invoked as one of the potential mechanisms for the beneficial effects of NMB in moderate/severe ARDS (15).
In follow-up studies, the same group questioned whether the injurious effects of spontaneous breathing could be mitigated by using volume-controlled ventilation (VCV) and limiting transpulmonary pressures (63). VCV, in contrast to pressure-controlled ventilation (PCV), places a constraint on the amount of VT inspired in the setting of strong spontaneous effort. Thus, they reasoned that VCV with strong spontaneous effort would be lung protective by limiting transpulmonary pressure, relative to PCV with strong spontaneous effort. Surfactant-depleted rabbits were subject to moderate (approximately 8 mL/kg) VT and low (3 cmH2O) PEEP in either VCV or PCV with and without spontaneous effort. While VCV was successful at reducing transpulmonary pressure, it was not successful at preventing damage. CT scans demonstrated occult pendelluft during strong spontaneous effort in dependent regions of the lung, causing regional over-distension. These findings were confirmed in a parallel porcine model of surfactant depletion using electrical impedance tomography (EIT), which showed movement of air from non-dependent to dependent lung regions (i.e., pendelluft) during spontaneous respiration. In a follow-up study (64), using a model of severe ALI (surfactant depletion followed by injurious ventilation), the damaging effects of pendelluft and dependent hyperinflation during spontaneous effort were largely abrogated by using higher PEEP (13 versus 5 cmH2O). In these experiments, higher PEEP resulted in lower transpulmonary pressure by reducing strong inspiratory drive (thus mitigating a strongly negative pleural pressure with spontaneous effort) and with more uniform redistribution of the VT over more recruited lung. Thus, high enough levels of PEEP can potentially permit the use of spontaneous breathing, even in severe ARDS, by minimizing pendelluft and regional overdistension in the dependent lung regions. However, this requires an explicit trial comparing NMB to a high PEEP strategy, as well as an assessment of both short- and long-term outcomes.
These findings complement the previously discussed finding of atelectasis and overdistension occurring in close proximity to one another using hyperpolarized gas MRI (44) on a global, rather than local, scale. The novel use of CT and EIT reinforce the utility of imaging to identify regional mechanics as a supplement to global mechanics when understanding the risk factors and mechanisms of VILI.
Pathways implicated in ARDS
Multiple inflammatory pathways have been implicated in ARDS on the basis of animal models [reviewed in (65,66)]. A thorough discussion of all implicated pathways is beyond the scope of this manuscript. Indeed, the lack of translation of many of these therapies to human ARDS, despite promise in animal ALI, should prompt us to re-assess how well we actually understand the pathophysiology of this syndrome, and to carefully consider the steps between pathway identification in an animal model to trials in humans. We will focus on two pathways which have received greater attention in recent studies as illustrative.
Inflammasome
Inflammasomes are intracellular protein complexes activated during infection or stress which serve as a platform for activation of caspase-1, which in turn activates the interleukin (IL)-1 family cytokines IL-1β and IL-18 (67). After demonstrating elevated levels of circulating IL-1β and IL-18 in subjects with ARDS, a group of investigators assessed the relevance of the inflammasome in a mouse model of VILI (VT 12 mL/kg for 8 hours) (68). In this model, VILI induced elevated inflammasome cytokines, with the alveolar macrophage as the likely origin. Genetic deletion of either IL-18 or caspase-1 conferred resistance to VILI, as did pharmacologic inhibition of IL-18. The relevance of the inflammasome was confirmed in separate model of LPS + VILI, in which genetic deletion of caspase-1 and pharmacologic inhibition of IL-1β protected mice from hypoxemia (69). Deletion of caspase-1 also protected mice subject to intratracheal LPS (70). Finally, pharmacologic IL-1β inhibition has been demonstrated to protect rats from VILI (28).
Importantly, all of these models implying benefit to neutralization of the inflammasome or its cytokines were sterile, and did not involve live bacteria. In a mouse pneumonia model using Pseudomonas aeruginosa, induction of the inflammasome was associated with impaired bacterial clearance and increased mortality (71). Depletion of alveolar macrophages, inhibition of caspase-1, or genetic deletion of IL-1β or IL-18 improved bacterial clearance and mortality, suggesting a therapeutic role for inflammasome inhibition even in an ALI model with live bacteria.
A separate investigation investigating the role of Staphylococcus aureus alpha toxin provides some additional nuance to these findings (72). In this mouse pneumonia model, administration of Staphylococcus or alpha toxin was sufficient to activate the inflammasome, and inflammasome activation prevented bacterial clearance. However, inhibition of neither IL-1β, IL-18, nor both improved bacterial clearance and mortality. However, direct inhibition of the inflammasome itself or caspase-1 improved bacterial clearance and mortality. This was affected via re-direction of cellular mitochondria away from inflammasomes and towards phagosomes containing bacteria. This effect was mediated by alpha toxin. In other words, Staphylococcus aureus alpha toxin induces inflammasome activation, which draws mitochondria towards the inflammasome and away from bacteria-containing phagosomes, thus preventing bacterial clearance. In this scenario, inflammasome inhibition, but not cytokine inhibition, improved bacterial clearance and outcomes. As pneumonia is the most common cause of ARDS, premature trials of IL-1β or IL-18 inhibition in human ARDS may have been doomed to failure, due to an imprecise appreciation of the underlying pathophysiology. These studies also highlight the importance of testing ALI therapies in multiple models, some of which need to include live bacteria, given the prevalence of infection as an inciting etiology in human ARDS.
Soluble receptor for advanced glycation end-products
The soluble receptor for advanced glycation end-products (sRAGE) has long been identified as a biomarker of ARDS diagnosis and prognosis (73-77). RAGE is a membrane receptor most highly expressed on type I alveolar epithelial cells, but also expressed in other tissues. RAGE is a pattern recognition receptor activated by AGEs. The soluble form (sRAGE), resulting from either alternative splicing or by active cleavage, has been suggested to be an epithelial-specific marker of ARDS severity, and elevated sRAGE is consistently associated with poor outcome in ARDS. The source and function of sRAGE is not entirely clear, and it is possible that it is a marker of epithelial damage; alternatively, in the setting of inflammation, elevated sRAGE may act as a decoy receptor to dampen inflammation by binding AGEs and other agonists of membrane-bound RAGE.
In a model of acid aspiration in which mice were allowed to recover, levels of alveolar and plasma sRAGE rose initially after injury and returned to baseline by day 4, tracking with changes in oxygenation and permeability (78). Alveolar fluid clearance, measured by instilling a tracer protein intratracheally and measuring its presence at subsequent timepoints, was impaired during ALI, and was inversely correlated with sRAGE levels. Thus, these authors concluded that elevated sRAGE was associated impaired alveolar fluid clearance during ALI. These findings were validated in a small cohort of humans with ARDS.
In a follow-up study, these same authors investigated the effects of modulating the RAGE axis in an acid aspiration model of ALI in mice (79). Treatment with either sRAGE or a monoclonal antibody inhibiting membrane-bound RAGE prevented hypoxemia, restored alveolar fluid clearance, and attenuated alveolar permeability after acid aspiration. Separate studies have demonstrated the benefits of RAGE inhibition in a mouse pneumonia model (80), suggesting that modulating this pathway may have some utility in ALI models with live bacteria, as well. Finally, the similar results seen from treatment with sRAGE or an antibody inhibiting the receptor suggests that sRAGE may be functioning primarily as a decoy receptor during ARDS, which is why it correlates well with epithelial damage, alveolar fluid clearance, ARDS severity, and mortality.
Conclusions
Despite the challenges of animal models in studying ARDS, significant advances have been made. Future studies should more consistently evaluate the regional heterogeneity of ARDS, with use of novel and more sensitive imaging modalities to assess regional, in addition to, global mechanics. Therapeutics should likely be tested in multiple models, as none sufficiently reproduce the heterogeneity of ARDS. This includes direct versus indirect ALI models, as well as those with and without live bacteria. Two-hit models may more accurately reproduce human ARDS, although their use requires additional skill. Better and longer supportive care mimicking the intensive care unit, particularly for larger animal models which can withstand ventilation for >8 hours, are also necessary for improved translation to humans. As the clinical definition of ARDS improves, and biomarker-defined subtypes of disease are identified, these will need to be explicitly modeled, as well. Ongoing interaction between clinicians, basic scientists, and translational scientists is essential for future optimal design of future ARDS studies.
Acknowledgments
Funding: This study is supported by the funding (NIH K23HL136688).
Footnote
Conflicts of Interest: The author has no conflicts of interest to declare.
Ethical Statement: The author is accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
References
- Ashbaugh DG, Bigelow DB, Petty TL, et al. Acute respiratory distress in adults. Lancet 1967;2:319-23. [Crossref] [PubMed]
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000;342:1334-49. [Crossref] [PubMed]
- Force ADT, Ranieri VM, Rubenfeld GD, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA 2012;307:2526-33. [PubMed]
- Beasley MB. The pathologist's approach to acute lung injury. Arch Pathol Lab Med 2010;134:719-27. [PubMed]
- Thille AW, Esteban A, Fernandez-Segoviano P, et al. Comparison of the Berlin definition for acute respiratory distress syndrome with autopsy. Am J Respir Crit Care Med 2013;187:761-7. [Crossref] [PubMed]
- Ashbaugh DG, Maier RV. Idiopathic pulmonary fibrosis in adult respiratory distress syndrome. Diagnosis and treatment. Arch Surg 1985;120:530-5. [Crossref] [PubMed]
- Herridge MS, Cheung AM, Tansey CM, et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med 2003;348:683-93. [Crossref] [PubMed]
- Bellani G, Laffey JG, Pham T, et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016;315:788-800. [Crossref] [PubMed]
- Amato MB, Meade MO, Slutsky AS, et al. Driving pressure and survival in the acute respiratory distress syndrome. N Engl J Med 2015;372:747-55. [Crossref] [PubMed]
- Acute Respiratory Distress Syndrome Network, Brower RG, Matthay MA, et al. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000;342:1301-8. [Crossref] [PubMed]
- National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network, Wiedemann HP, Wheeler AP, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 2006;354:2564-75.
- National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network, Matthay MA, Brower RG, et al. Randomized, placebo-controlled clinical trial of an aerosolized beta(2)-agonist for treatment of acute lung injury. Am J Respir Crit Care Med 2011;184:561-8.
- Rice TW, Wheeler AP, Thompson BT, et al. Enteral omega-3 fatty acid, gamma-linolenic acid, and antioxidant supplementation in acute lung injury. JAMA 2011;306:1574-81. [Crossref] [PubMed]
- Steinberg KP, Hudson LD, Goodman RB, et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med 2006;354:1671-84. [Crossref] [PubMed]
- Papazian L, Forel JM, Gacouin A, et al. Neuromuscular blockers in early acute respiratory distress syndrome. N Engl J Med 2010;363:1107-16. [Crossref] [PubMed]
- Guerin C, Reignier J, Richard JC, et al. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med 2013;368:2159-68. [Crossref] [PubMed]
- Combes A, Hajage D, Capellier G, et al. Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Distress Syndrome. N Engl J Med 2018;378:1965-75. [Crossref] [PubMed]
- Meade MO, Cook DJ, Guyatt GH, et al. Ventilation strategy using low tidal volumes, recruitment maneuvers, and high positive end-expiratory pressure for acute lung injury and acute respiratory distress syndrome: a randomized controlled trial. JAMA 2008;299:637-45. [Crossref] [PubMed]
- Mercat A, Richard JC, Vielle B, et al. Positive end-expiratory pressure setting in adults with acute lung injury and acute respiratory distress syndrome: a randomized controlled trial. JAMA 2008;299:646-55. [Crossref] [PubMed]
- Goligher EC, Kavanagh BP, Rubenfeld GD, et al. Oxygenation response to positive end-expiratory pressure predicts mortality in acute respiratory distress syndrome. A secondary analysis of the LOVS and ExPress trials. Am J Respir Crit Care Med 2014;190:70-6. [Crossref] [PubMed]
- Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 2008;295:L379-99. [Crossref] [PubMed]
- Schneemann M, Schoedon G. Species differences in macrophage NO production are important. Nat Immunol 2002;3:102. [Crossref] [PubMed]
- Rehli M. Of mice and men: species variations of Toll-like receptor expression. Trends Immunol 2002;23:375-8. [Crossref] [PubMed]
- Sone Y, Serikov VB, Staub NC Sr. Intravascular macrophage depletion attenuates endotoxin lung injury in anesthetized sheep. J Appl Physiol (1985) 1999;87:1354-9. [PubMed]
- Matute-Bello G, Downey G, Moore BB, et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 2011;44:725-38. [Crossref] [PubMed]
- Uhlig S, Uhlig U. Pharmacological interventions in ventilator-induced lung injury. Trends Pharmacol Sci 2004;25:592-600. [Crossref] [PubMed]
- Tsuno K, Miura K, Takeya M, et al. Histopathologic pulmonary changes from mechanical ventilation at high peak airway pressures. Am Rev Respir Dis 1991;143:1115-20. [Crossref] [PubMed]
- Frank JA, Pittet JF, Wray C, et al. Protection from experimental ventilator-induced acute lung injury by IL-1 receptor blockade. Thorax 2008;63:147-53. [Crossref] [PubMed]
- Ricard JD, Dreyfuss D, Saumon G. Production of inflammatory cytokines in ventilator-induced lung injury: a reappraisal. Am J Respir Crit Care Med 2001;163:1176-80. [Crossref] [PubMed]
- Dries DJ, Adams AB, Marini JJ. Time course of physiologic variables in response to ventilator-induced lung injury. Respir Care 2007;52:31-7. [PubMed]
- de Prost N, Roux D, Dreyfuss D, et al. Alveolar edema dispersion and alveolar protein permeability during high volume ventilation: effect of positive end-expiratory pressure. Intensive Care Med 2007;33:711-7. [Crossref] [PubMed]
- Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis 1974;110:556-65. [PubMed]
- Parker JC, Hernandez LA, Longenecker GL, et al. Lung edema caused by high peak inspiratory pressures in dogs. Role of increased microvascular filtration pressure and permeability. Am Rev Respir Dis 1990;142:321-8. [Crossref] [PubMed]
- Carlton DP, Cummings JJ, Scheerer RG, et al. Lung overexpansion increases pulmonary microvascular protein permeability in young lambs. J Appl Physiol (1985) 1990;69:577-83. [PubMed]
- Egan EA, Nelson RM, Olver RE. Lung inflation and alveolar permeability to non-electrolytes in the adult sheep in vivo. J Physiol 1976;260:409-24. [Crossref] [PubMed]
- Egan EA. Response of alveolar epithelial solute permeability to changes in lung inflation. J Appl Physiol Respir Environ Exerc Physiol 1980;49:1032-6. [PubMed]
- Wilson MR, Patel BV, Takata M. Ventilation with "clinically relevant" high tidal volumes does not promote stretch-induced injury in the lungs of healthy mice. Crit Care Med 2012;40:2850-7. [Crossref] [PubMed]
- Caironi P, Langer T, Carlesso E, et al. Time to generate ventilator-induced lung injury among mammals with healthy lungs: a unifying hypothesis. Intensive Care Med 2011;37:1913-20. [Crossref] [PubMed]
- Dreyfuss D, Soler P, Basset G, et al. High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis 1988;137:1159-64. [Crossref] [PubMed]
- Bshouty Z, Ali J, Younes M. Effect of tidal volume and PEEP on rate of edema formation in in situ perfused canine lobes. J Appl Physiol (1985) 1988;64:1900-7. [PubMed]
- Luce JM. The cardiovascular effects of mechanical ventilation and positive end-expiratory pressure. JAMA 1984;252:807-11. [Crossref] [PubMed]
- Dreyfuss D, Saumon G. Role of tidal volume, FRC, and end-inspiratory volume in the development of pulmonary edema following mechanical ventilation. Am Rev Respir Dis 1993;148:1194-203. [Crossref] [PubMed]
- Pourfathi M, Cereda M, Chatterjee S, et al. Lung Metabolism and Inflammation during Mechanical Ventilation; An Imaging Approach. Sci Rep 2018;8:3525. [Crossref] [PubMed]
- Cereda M, Emami K, Xin Y, et al. Imaging the interaction of atelectasis and overdistension in surfactant-depleted lungs. Crit Care Med 2013;41:527-35. [Crossref] [PubMed]
- Halter JM, Steinberg JM, Gatto LA, et al. Effect of positive end-expiratory pressure and tidal volume on lung injury induced by alveolar instability. Crit Care 2007;11:R20. [Crossref] [PubMed]
- Brower RG, Lanken PN, MacIntyre N, et al. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med 2004;351:327-36. [Crossref] [PubMed]
- Writing Group for the Alveolar Recruitment for Acute Respiratory Distress Syndrome Trial I, Cavalcanti AB, Suzumura EA, et al. Effect of Lung Recruitment and Titrated Positive End-Expiratory Pressure (PEEP) vs Low PEEP on Mortality in Patients With Acute Respiratory Distress Syndrome: A Randomized Clinical Trial. JAMA 2017;318:1335-45. [Crossref] [PubMed]
- Stapleton RD, Wang BM, Hudson LD, et al. Causes and timing of death in patients with ARDS. Chest 2005;128:525-32. [Crossref] [PubMed]
- Dowell JC, Parvathaneni K, Thomas NJ, et al. Epidemiology of Cause of Death in Pediatric Acute Respiratory Distress Syndrome. Crit Care Med 2018;46:1811-9. [Crossref] [PubMed]
- Fain SB, Korosec FR, Holmes JH, et al. Functional lung imaging using hyperpolarized gas MRI. J Magn Reson Imaging 2007;25:910-23. [Crossref] [PubMed]
- Cereda M, Emami K, Kadlecek S, et al. Quantitative imaging of alveolar recruitment with hyperpolarized gas MRI during mechanical ventilation. J Appl Physiol (1985) 2011;110:499-511. [PubMed]
- Bayat S, Porra L, Albu G, et al. Effect of positive end-expiratory pressure on regional ventilation distribution during mechanical ventilation after surfactant depletion. Anesthesiology 2013;119:89-100. [Crossref] [PubMed]
- Cereda M, Xin Y, Meeder N, et al. Visualizing the Propagation of Acute Lung Injury. Anesthesiology 2016;124:121-31. [Crossref] [PubMed]
- Musch G, Bellani G, Vidal Melo MF, et al. Relation between shunt, aeration, and perfusion in experimental acute lung injury. Am J Respir Crit Care Med 2008;177:292-300. [Crossref] [PubMed]
- Richter T, Bellani G, Scott Harris R, et al. Effect of prone position on regional shunt, aeration, and perfusion in experimental acute lung injury. Am J Respir Crit Care Med 2005;172:480-7. [Crossref] [PubMed]
- Jones HA, Clark RJ, Rhodes CG, et al. In vivo measurement of neutrophil activity in experimental lung inflammation. Am J Respir Crit Care Med 1994;149:1635-9. [Crossref] [PubMed]
- Musch G, Venegas JG, Bellani G, et al. Regional gas exchange and cellular metabolic activity in ventilator-induced lung injury. Anesthesiology 2007;106:723-35. [Crossref] [PubMed]
- de Prost N, Costa EL, Wellman T, et al. Effects of surfactant depletion on regional pulmonary metabolic activity during mechanical ventilation. J Appl Physiol (1985) 2011;111:1249-58. [PubMed]
- Pourfathi M, Xin Y, Kadlecek SJ, et al. In vivo imaging of the progression of acute lung injury using hyperpolarized [1-(13) C] pyruvate. Magn Reson Med 2017;78:2106-15. [Crossref] [PubMed]
- Xia J, Sun B, He H, et al. Effect of spontaneous breathing on ventilator-induced lung injury in mechanically ventilated healthy rabbits: a randomized, controlled, experimental study. Crit Care 2011;15:R244. [Crossref] [PubMed]
- Yoshida T, Uchiyama A, Matsuura N, et al. Spontaneous breathing during lung-protective ventilation in an experimental acute lung injury model: high transpulmonary pressure associated with strong spontaneous breathing effort may worsen lung injury. Crit Care Med 2012;40:1578-85. [Crossref] [PubMed]
- Yoshida T, Uchiyama A, Matsuura N, et al. The comparison of spontaneous breathing and muscle paralysis in two different severities of experimental lung injury. Crit Care Med 2013;41:536-45. [Crossref] [PubMed]
- Yoshida T, Nakahashi S, Nakamura MAM, et al. Volume-controlled Ventilation Does Not Prevent Injurious Inflation during Spontaneous Effort. Am J Respir Crit Care Med 2017;196:590-601. [Crossref] [PubMed]
- Morais CCA, Koyama Y, Yoshida T, et al. High Positive End-Expiratory Pressure Renders Spontaneous Effort Noninjurious. Am J Respir Crit Care Med 2018;197:1285-96. [Crossref] [PubMed]
- Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 2012;122:2731-40. [Crossref] [PubMed]
- Reiss LK, Schuppert A, Uhlig S. Inflammatory processes during acute respiratory distress syndrome: a complex system. Curr Opin Crit Care 2018;24:1-9. [Crossref] [PubMed]
- Schroder K, Tschopp J. The inflammasomes. Cell 2010;140:821-32. [Crossref] [PubMed]
- Dolinay T, Kim YS, Howrylak J, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med 2012;185:1225-34. [Crossref] [PubMed]
- Jones HD, Crother TR, Gonzalez-Villalobos RA, et al. The NLRP3 inflammasome is required for the development of hypoxemia in LPS/mechanical ventilation acute lung injury. Am J Respir Cell Mol Biol 2014;50:270-80. [PubMed]
- Grailer JJ, Canning BA, Kalbitz M, et al. Critical role for the NLRP3 inflammasome during acute lung injury. J Immunol 2014;192:5974-83. [Crossref] [PubMed]
- Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest 2013;123:1630-7. [Crossref] [PubMed]
- Cohen TS, Boland ML, Boland BB, et al. S. aureus Evades Macrophage Killing through NLRP3-Dependent Effects on Mitochondrial Trafficking. Cell Rep 2018;22:2431-41. [Crossref] [PubMed]
- Jabaudon M, Futier E, Roszyk L, et al. Soluble form of the receptor for advanced glycation end products is a marker of acute lung injury but not of severe sepsis in critically ill patients. Crit Care Med 2011;39:480-8. [Crossref] [PubMed]
- Liu X, Chen Q, Shi S, et al. Plasma sRAGE enables prediction of acute lung injury after cardiac surgery in children. Crit Care 2012;16:R91. [Crossref] [PubMed]
- Matsumoto H, Matsumoto N, Ogura H, et al. The clinical significance of circulating soluble RAGE in patients with severe sepsis. J Trauma Acute Care Surg 2015;78:1086-93; discussion 1093-4. [PubMed]
- Yehya N, Thomas NJ, Meyer NJ, et al. Circulating markers of endothelial and alveolar epithelial dysfunction are associated with mortality in pediatric acute respiratory distress syndrome. Intensive Care Med 2016;42:1137-45. [Crossref] [PubMed]
- Mrozek S, Jabaudon M, Jaber S, et al. Elevated Plasma Levels of sRAGE Are Associated With Nonfocal CT-Based Lung Imaging in Patients With ARDS: A Prospective Multicenter Study. Chest 2016;150:998-1007. [Crossref] [PubMed]
- Jabaudon M, Blondonnet R, Roszyk L, et al. Soluble Receptor for Advanced Glycation End-Products Predicts Impaired Alveolar Fluid Clearance in Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 2015;192:191-9. [Crossref] [PubMed]
- Blondonnet R, Audard J, Belville C, et al. RAGE inhibition reduces acute lung injury in mice. Sci Rep 2017;7:7208. [Crossref] [PubMed]
- Christaki E, Opal SM, Keith JC Jr, et al. A monoclonal antibody against RAGE alters gene expression and is protective in experimental models of sepsis and pneumococcal pneumonia. Shock 2011;35:492-8. [Crossref] [PubMed]

